iSeqQC- An Expression based Quality Control tool
Welcome to iSeqQC
It is an expression-based quality control tool to detect outliers either produced by batch effects or merely due to dissimilarity within a phenotypic group.
Input: 1) Choose the file-type between raw or normalized counts options;
2) Choose the organism of study;
3) Choose between gene symbol or gene id as in your first column of counts file
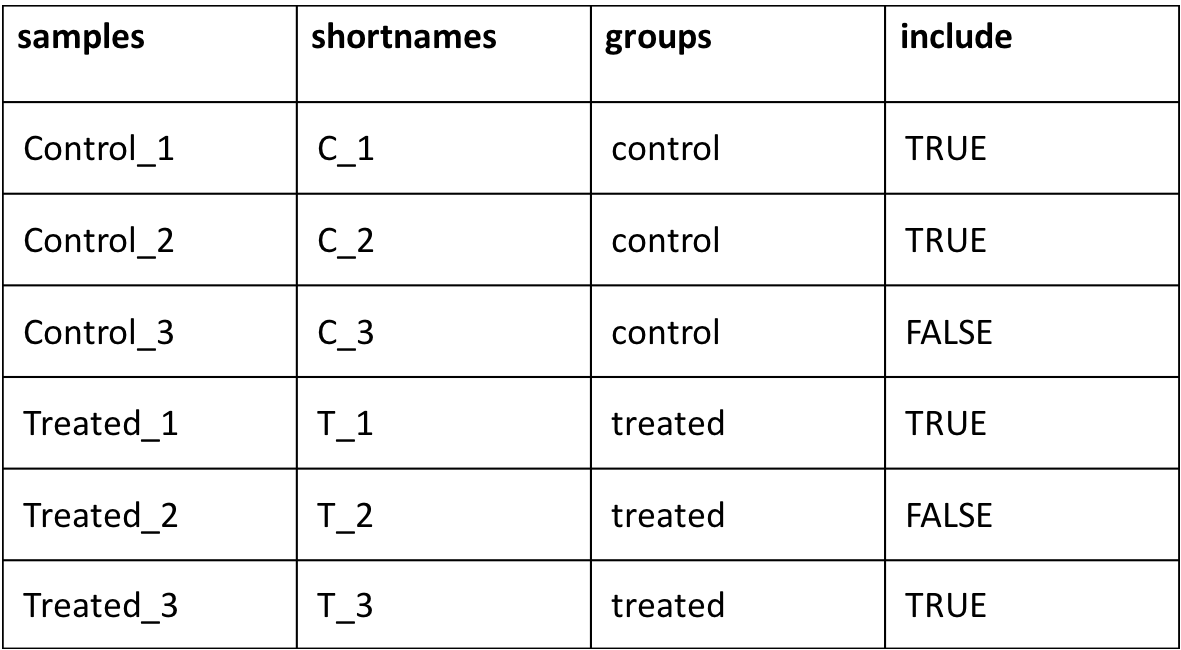
4) Sample phenotype file in tab-delimited with a minimum of 4 columns, 1) sample names; 2) short name of samples; 3) groups; 4) samples to include in the analysis; 5) any factors that could influence any bias such as library protocol or preparation method. The first 4 columns are mandatory and should strictly match the names and order as mentioned below (names case-sensitive), column 5-11 are optional and could be left blank
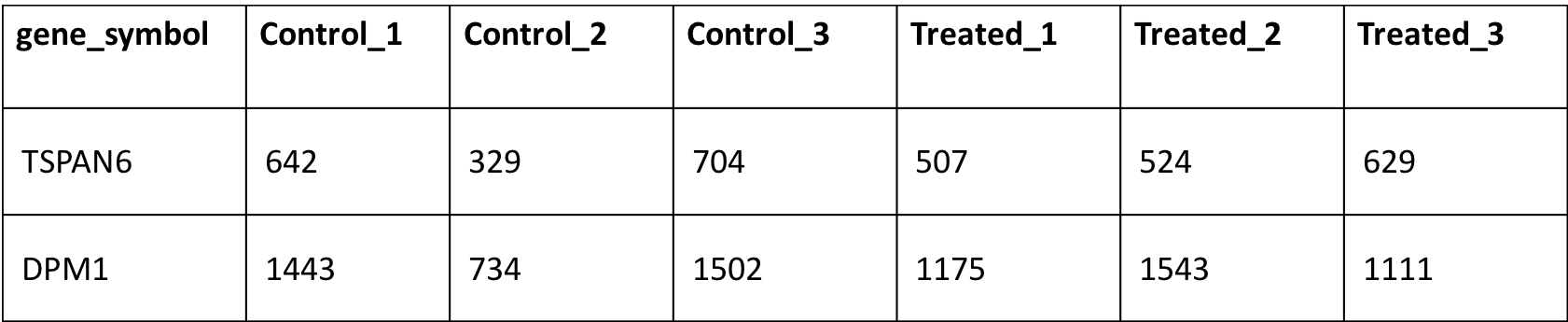
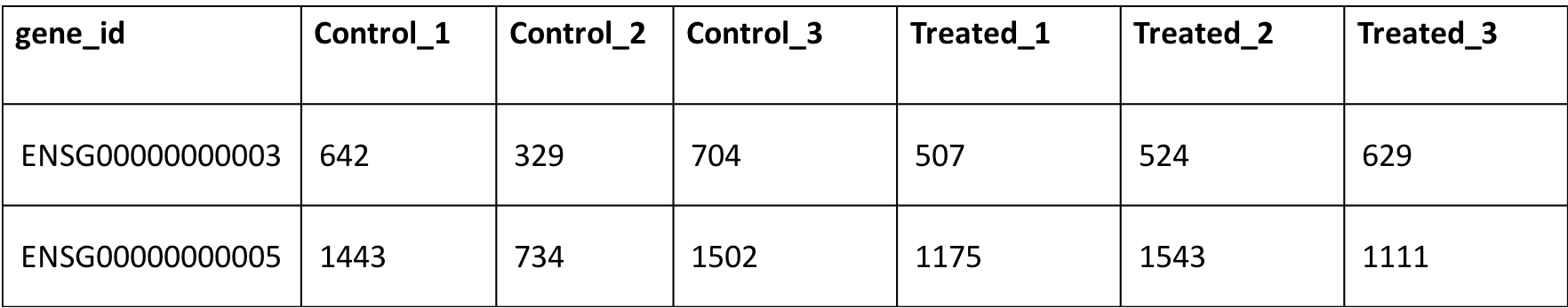
5) A counts matrix file in tab-delimited file. This could be a raw or normalized (TPM/FPKM/log2/RPKM/RPM etc.). This file should have gene identifier (either gene symbol or gene id's) in first column as shown below
or
Output: It is presented in two tabs, where in ‘Quality Control’, you are required to input the above-mentioned information and obtain output is in form of an expression summary table and several QC plots such as density plots, PCA, hierchichal clustering, correlation plots. In ‘Expression Plots’ tab, you can choose your gene of interest to plot expression profile among different phenotypic groups.
Please note: To remove any sample from analysis, simply replace 'TRUE' with 'FALSE' in 'include' column in sample manifest file.
Additional information can be obtained at https://github.com/gkumar09/iSeqQC
Citation:
Kumar G, Ertel A, Feldman G, Kupper J, Fortina P (2020). iSeqQC: A Tool for Expression-Based Quality Control in RNA Sequencing. BMC Bioinformatics. Feb 13;21(1):56. doi: 10.1186/s12859-020-3399-8. PMID: 32054449; PMCID: PMC7020508
Contact:
Gaurav Kumar, PhD
Email address: gaurav.kumar@jefferson.edu
It is an expression-based quality control tool to detect outliers either produced by batch effects or merely due to dissimilarity within a phenotypic group.
Input: 1) Choose the file-type between raw or normalized counts options;
2) Choose the organism of study;
3) Choose between gene symbol or gene id as in your first column of counts file
4) Sample phenotype file in tab-delimited with a minimum of 4 columns, 1) sample names; 2) short name of samples; 3) groups; 4) samples to include in the analysis; 5) any factors that could influence any bias such as library protocol or preparation method. The first 4 columns are mandatory and should strictly match the names and order as mentioned below (names case-sensitive), column 5-11 are optional and could be left blank
5) A counts matrix file in tab-delimited file. This could be a raw or normalized (TPM/FPKM/log2/RPKM/RPM etc.). This file should have gene identifier (either gene symbol or gene id's) in first column as shown below
or
Output: It is presented in two tabs, where in ‘Quality Control’, you are required to input the above-mentioned information and obtain output is in form of an expression summary table and several QC plots such as density plots, PCA, hierchichal clustering, correlation plots. In ‘Expression Plots’ tab, you can choose your gene of interest to plot expression profile among different phenotypic groups.
Please note: To remove any sample from analysis, simply replace 'TRUE' with 'FALSE' in 'include' column in sample manifest file.
Additional information can be obtained at https://github.com/gkumar09/iSeqQC
Citation:
Kumar G, Ertel A, Feldman G, Kupper J, Fortina P (2020). iSeqQC: A Tool for Expression-Based Quality Control in RNA Sequencing. BMC Bioinformatics. Feb 13;21(1):56. doi: 10.1186/s12859-020-3399-8. PMID: 32054449; PMCID: PMC7020508
Contact:
Gaurav Kumar, PhD
Email address: gaurav.kumar@jefferson.edu
Download the distribution of counts per sample
Download the Mapped Reads density per sample
Download the housekeeping genes expression per sample
Download the Principal components plot for all samples- normalized
Download the Principal components plot for all samples- non-normalized
Download the Multifactor Principal components plot
Download the hierarchichal plot
Download the pearson correlation plot
Download the spearman correlation plot
Download the GC bias plot